Introduction
The SARS-CoV-2 pandemic has generated a demand for sequencing-based viral genome analysis at an unprecedented scale. As a result of this challenge, numerous new sequencing projects have emerged to track the pandemic on a molecular level. Fortunately, investments made in public and open computing and storage infrastructure projects, as well as in reliable and scalable data analysis frameworks have proven beneficial and could be employed for the required genome monitoring of SARS-CoV-2.
Here, we describe a modular and scalable system for FAIR analysis of SARS-CoV-2 sequencing data. This effort shows how the Galaxy Project with the help of many additional collaborators repurposed, expanded, and combined public infrastructure and systems for storing and sharing data, and for managing data analysis workflows, to tackle a global health emergency.
Who is this showcase intended for?
Automated Galaxy workflow runs for viral genome surveillance are of interest for any department, institution or organisation that intend to perform routine genome monitoring of virus sequences at non-trivial scale, and who care about FAIR large-scale data analysis.
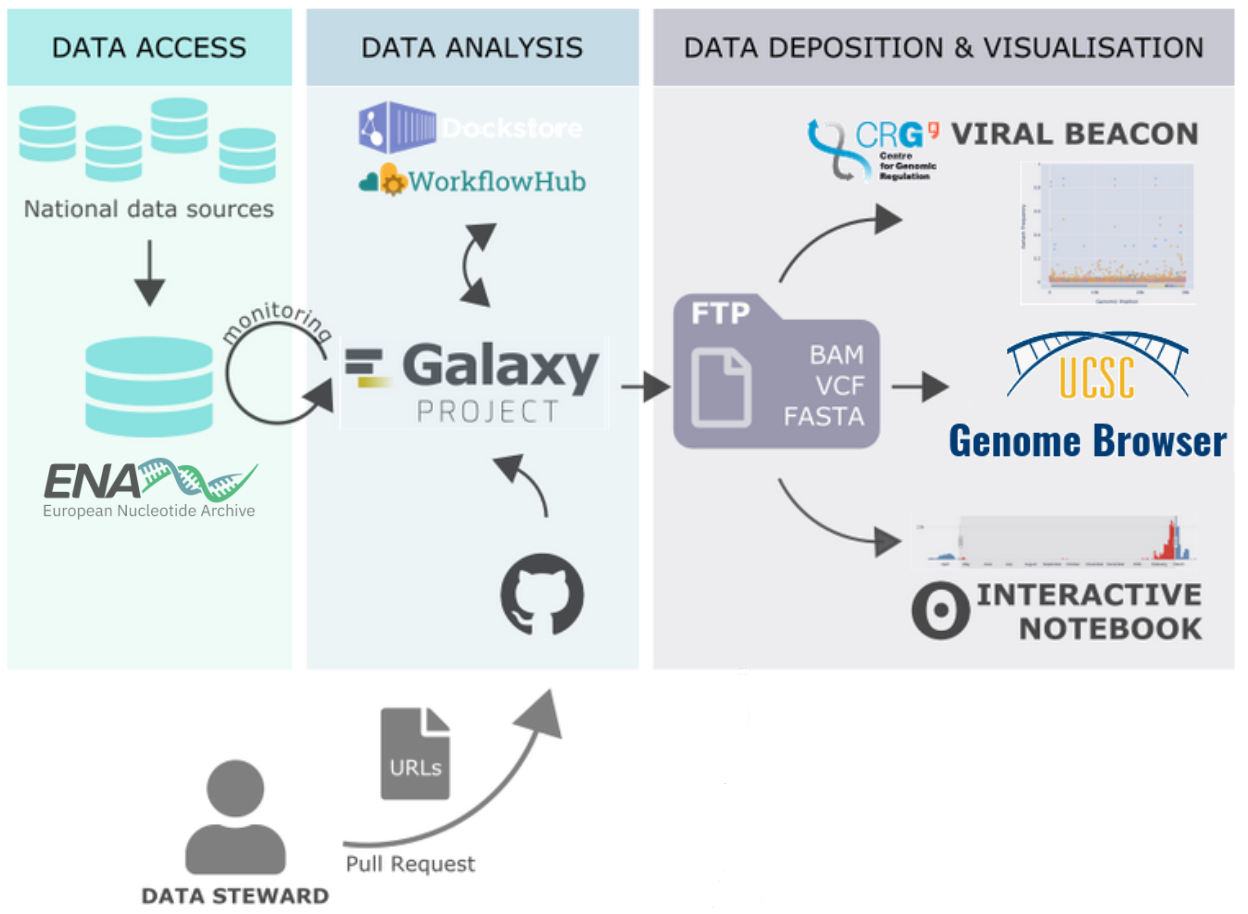
Figure 1. An automated SARS-CoV-2 sequencing data analysis system built around Galaxy as the analysis engine. Data to process is discovered and pulled into Galaxy from the European Nucleotide Archive (ENA) and processed via versioned releases of Galaxy Workflows published at and downloaded via WorkflowHub and/or Dockstore. Third parties (data stewards) can also suggest their own public samples of interest to be included in analysis runs by opening a pull request on GitHub. Key files for every analysis run are exported to a publicly accessible archive provided by the Centre of Genomic Regulation (CRG), and can be consumed by downstream visualisations like CRG’s Viral Beacon, the UCSC genome browser, and custom interactive notebooks.
Building blocks
European Nucleotide Archive (ENA)
Many large national SARS-CoV-2 sequencing data providers submit their raw data to the European Nucleotide Archive (ENA). In the showcase we are using the ENA’s public API to extract links to the raw sequencing data of newly submitted sequenced reads from several large-scale national genome surveillance efforts including (but not limited to):
- the COVID-19 Genomics UK Consortium (COG-UK; ENA Project accession: PRJEB37886)
- the Portuguese network for SARS-CoV-2 genomics (ENA Project accession: PRJEB47340)
- the Estonian national sequencing initiatives (KoroGeno-EST-3 and KoroGeno-EST-2022; see dedicated showcase)
In addition, we provide the possibility to request analysis of samples of particular interest via pull requests against a dedicated GitHub repository.
WorkflowHub
The WorkflowHub registry for scientific computational workflows provides access to hundreds of workflows with defined releases, among them an evolving set of Galaxy workflows for SARS-CoV-2 genome analysis.
These are provided and/or reviewed by the IWC, a subgroup within the Galaxy Community concerned with maintaining high-quality Galaxy Workflows and who collaborates closely with WorkflowHub on FAIR workflow matters.
Most IWC SARS-CoV-2 analysis workflows follow a modular design decision, i.e. there are separate workflows for discovering viral mutations from raw sequencing data obtained from different sequencing platforms and protocols, for reporting and visualising these mutations within and across samples, and for generating viral consensus genomes.
This design provides users with the advantage of being able to combine analysis modules according to their specific needs.
Galaxy Europe
The Galaxy Europe server provides free access to powerful publicly-funded compute infrastructure and thousands of bioinformatics tools.
In the showcase we download ENA-hosted or user-requested sequencing data to the Galaxy server and process it with the workflows from WorkflowHub.
Archive
The archive is provided by the Centre for Genomic Regulation (CRG).
For every sample processed through Galaxy workflow runs, we are exporting key results files (reads mapped to the viral reference sequence, variant calls, consensus sequences and tabular reports) to a public archive so that consumers can discover, access and reuse it.
We are also maintaining a provenance JSON file in this archive that makes it possible to reconstruct the full data processing history of each sample.
You can access all the data by logging in to: ftp://xfer13.crg.eu as User: FTPuser Password: FTPusersPassword
Viral Beacon project
In the showcase the CRG COVID-19 Viral Beacon acts as a consumer of the data and provides visualisations.
UCSC genome browser
The UCSC Genome Browser offers a Galaxy ENA mutations track populated from the data on the CRG’s archive.
Bringing it all together
The components are connected using free and open source “glue” code that can be found on GitHub.
The code relies, to a large extent, on built-in Galaxy functionality, which it accesses via the Galaxy API. This approach allows us to keep the code minimal.
The automation code takes care of orchestrating the steps of
- uploading ENA data via URLs
- running all workflows necessary for an analysis of a given sample batch from the set offered on WorkflowHub in an automated manner
- sending result datasets to the remote archive
- tagging analysis artefacts for easier discovery, publishing analyses for accessibility, etc.
What can you use this showcase for?
This showcase illustrates how you can combine existing public frameworks, registries, and data repositories, that have all been created or enhanced significantly over the course of the COVID-19 pandemic, to conduct automated, reproducible, shareable, transparent, high-quality viral sequencing data analysis at any scale using open-source code.
The system was designed with a broad range of users in mind:
- Any researcher can reuse any of the archived result files produced by the showcase project for retrospective downstream analyses.
- Virologists can use the Github-based analysis on request feature to get a high-quality, reliable analysis of their own samples performed on the Galaxy Europe server.
- Genome surveillance initiatives can reuse the entire system on-premises or exchange components as they see fit.
How to reuse the components
An important aspect of the system presented here in terms of reusability is its modular architecture. You could for example:
-
Change the Galaxy server instance used to process the data
All analysis tools used in the Galaxy workflows are publicly available and are easy to install on any, public or private, Galaxy instance.
While it is easiest for occasional users to just use the Github-based analysis request feature to have their data processed by Galaxy Europe, larger genome surveillance projects may want to run the system on their own dedicated instance of Galaxy to enjoy shorter queue times and to ensure privacy of the data before its release to public archives.
-
Get the raw sequencing data from sources other than the ENA
As long as you can give Galaxy access to the data there is a way to feed it into the system.
-
Build and run modified versions or combinations of the workflows
Galaxy comes with an easy-to-use graphical workflow editor that lets you adapt any of the existing public workflows to your specific needs, and the modular design of the workflows makes it rather straightforward to build the ideal pipeline for your purpose.
You can also simply clone the automation glue code and modify its config files to orchestrate your custom pipeline on your Galaxy instance of choice from a local computer or server.
-
Send result files to a different remote file system
Galaxy has a plugin system through which it can be connected to various remote file systems for data export.
-
Use the data to populate other dashboards of your choice
The Galaxy project and collaborators have, for example, come up with an interactive Observable dashboard as an alternative visualisation tool for the results produced as part of the showcase.
Acknowledgments
COVID19 Galaxy Project Team Members
Wolfgang Maier, Simon Bray, Anton Nekrutenko, Björn Grüning, Marius van den Beek and Dannon Baker from the Galaxy project; Babita Singh, Mauricio Moldes, Jordi Rambla from CRG and the Viral Beacon project; Maximilian Haeussler from the UCSC genome browser team; Sergei Pond (Temple University) developed interactive Observable notebooks.
Additional people who have helped improve the workflows used for the data analysis: Ulvi Talas (University of Tartu), Peter van Heusden (SANBI)
Support
Galaxy Europe is supported by de.NBI (the German Network for Bioinformatics Infrastructure) and through associated funding via the BMBF (German Federal Ministry of Education and Research) grants 031L0101C de.NBI-epi and 031 A538A de.NBI-RBC.
Related pages
Full-genomic sequencing and analysis of SARS-CoV-2 strains in Estonia during 2021-2022.
A web portal that provides information about available datasets, resources, tools, and services related to pandemic preparedness in Sweden. We offer support to all those involved in pandemic preparedness research that are affiliated with a Swedish research institution or university.
More information
Training
Tools and resources on this page
| Tool or resource | Description | Related pages | Registry |
|---|---|---|---|
| CRG COVID-19 Viral Beacon | A platform allowing for browsing SARS-CoV-2 variability at the genome, amino acid, structural, and motif levels | Human biomolecular data | |
| European Nucleotide Archive (ENA) | Provides a record of the nucleotide sequencing information. It includes raw sequencing data, sequence assembly information and functional annotation. | Pathogen characterisation Human clinical and hea... Pathogen characterisation Human biomolecular data Using the ENA data sub... SARS-CoV-2 sequencing ... Linked pathogen and ho... | Tool info Standards/Databases Training |
| Galaxy Europe | The European Galaxy server. Provides access to thousands of tools for scalable and reproducible analysis. | Pathogen characterisation | Training |
| UCSC Genome Browser | An online tool for analyzing and visualizing genomic data. It allows users to add and share annotations. | Human biomolecular data | Tool info Standards/Databases Training |
| WorkflowHub | A registry for describing, sharing and publishing scientific computational workflows. | Pathogen characterisation | Tool info Standards/Databases Training |


University of Freiburg / ELIXIR-DE